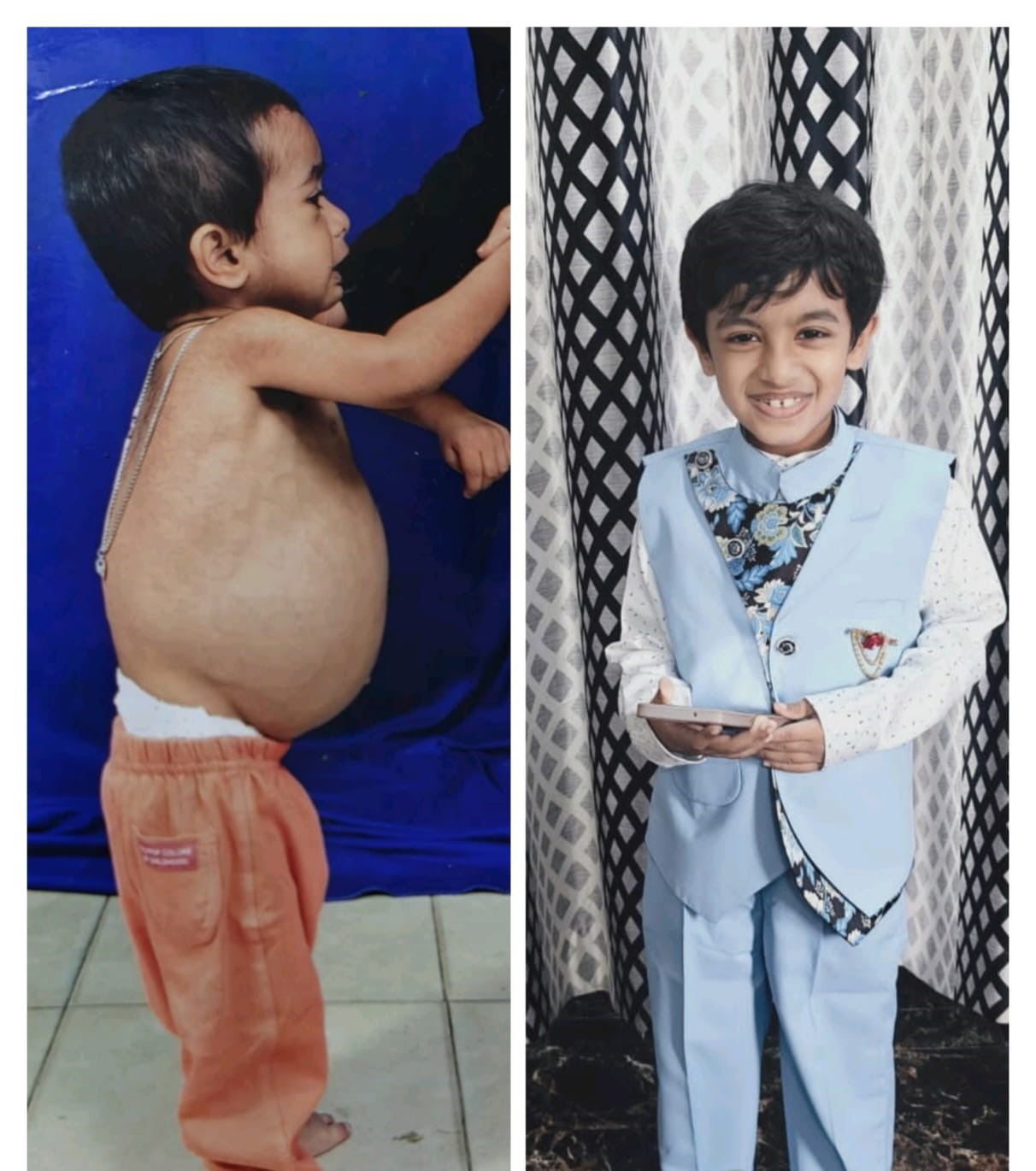
When Syed Arhab was just nine months old, his mother noticed something terrifying. Her baby's stomach was swelling unnaturally while his tiny arms and legs grew disturbingly thin. He stopped eating. What followed was every parent's nightmare; a cascade of medical tests, uncertain diagnoses, and finally, the devastating truth: Gaucher disease, a rare genetic disorder that would require lifelong treatment just to survive.
Today, Syed is eight years old. He should be playing cricket with friends, laughing in his first-grade classroom, and dreaming about his future. Instead, he's fighting for his life not because medicine has failed him, but because he cannot access the treatment that exists.
A Disease That Steals Childhood
Gaucher disease is one of the rarest genetic disorders in the world, affecting approximately one in 60,000 births. It occurs when the body cannot produce sufficient quantities of an enzyme called glucocerebrosidase, which normally breaks down fatty substances in cells. Without this enzyme, toxic lipids accumulate progressively in the liver, spleen, bone marrow, and sometimes the brain; creating what doctors call "Gaucher cells."
The results are devastating. Syed's liver and spleen became massively enlarged, causing the characteristic swollen belly that first alarmed his parents. His blood counts plummeted dangerously low. His bones weakened. He couldn't grow properly. He became too weak to stand; a toddler whose heavy, distended stomach overwhelmed his thinning limbs.
The cruelest part? A proven treatment exists. Enzyme replacement therapy (ERT) has been approved since 1991. Children who receive consistent ERT can live completely normal lives. They go to school, play sports, pursue careers, and have families of their own. The treatment works by replacing the missing enzyme through intravenous infusions every two weeks, gradually reversing the cellular destruction and restoring normal function.
But Syed cannot access this life-saving medicine.
Two Years of Darkness
When Syed was diagnosed at nine months, his parents were devastated. They didn't understand genetics or lysosomal storage disorders. They only understood that their baby was suffering, and the prognosis was grim without treatment. For two and a half agonizing years, Syed received nothing. Government hospitals couldn't procure the medication in time. Bureaucratic delays stretched into months. His symptoms worsened relentlessly. His stomach swelled further. His development stalled. He became too weak to walk.
His parents watched helplessly, knowing that somewhere in the world, the medicine existed that could save their son. But in their reality in a working-class family in Bangalore where his father sells furniture to support his wife and three children that medicine might as well have been on another planet.
A Glimpse of Hope
When Syed finally turned three, a miracle happened. The effort of Patient Advocacy group helped him to get enzyme replacement therapy. Within six months of starting treatment, the transformation was extraordinary. The swelling in his abdomen began to decrease. His strength started returning. For the first time in years, his parents saw their son smile without shadowing his eyes.
Syed began school. Against all odds, this resilient child who had suffered so profoundly started to live a normal life. He made friends. He learned to read. He showed the remarkable resilience that only children possess the ability to find joy even after unimaginable suffering.
But the treatment costs ₹3.5 lakhs every single month.
For a furniture shopkeeper supporting three children and a homemaker wife, this amount is impossible. Ten times their monthly income. An insurmountable wall between their son's survival and death.
The Current Crisis
The treatment became irregular, interrupted by funding crises that repeated every few months. Some months, Syed received his medication. Other months, the money simply wasn't there. Each interruption caused predictable, documented consequences: organ re-enlargement, declining blood counts, weakening strength.
In mid-2025, the worst happened. The hospital could no longer fund the treatment. Months passed without enzyme replacement therapy. The disease, given freedom to run unchecked, roared back with vengeance.
Seizures began, a terrifying complication affecting 30% of untreated Gaucher disease patients. He started coughing constantly. His appetite vanished again. He became too weak to focus on his studies or play like a normal eight-year-old.
Syed's last treatment was in June 2025. He has now gone five months without the medication keeping him alive.
His parents watch their son deteriorate, knowing exactly what he needs but powerless to provide it. His teenage sisters see their brother suffering and understand the fragility of life in ways no child should. The family is in agony.
Every day without treatment is a day of progressive cellular destruction, of accumulating toxic lipids poisoning his systems, of stolen opportunities and closing developmental windows.
Why Your Help Matters
This is not a story requiring experimental or unproven therapy. Enzyme replacement therapy for Gaucher disease is established standard of care, scientifically validated, and proven to work across thousands of patients worldwide.
What Syed needs is not a miracle, it is access to medicine the world has already developed and proven effective.
With consistent, uninterrupted ERT, there is every medical reason to expect Syed could:
- Recover from his current crisis within 3-6 months
- Return to normal childhood activities and school
- Grow into a healthy, functional adult
- Pursue any career he chooses
- Live a normal lifespan with quality of life